
MDD“ΫΝΤ…η±Η»œ÷Λ
ΜΣ―ΕΦλ≤β 2019-7-1“ΫΝΤΤς–Β»œ÷Λ93/42/EEC »œ÷ΛΝς≥ΧΦΑΗϋ–¬÷ΝMDR–¬÷ΗΝν±δΕ·
1: ≤Ο¥ «93/42/EEC“ΫΝΤΤς–Β÷ΗΝν?
÷ΗΝνΑϋά®ΝΥ“ΫΝΤ…η±Η“‘ΦΑΥϋΒΡ≈δΦΰ
ΗυΨί÷ΗΝν“ΫΝΤΤς–Β…η±Η“βΈΕΉ≈:
»ΈΚΈ“«ΤςΓΔΤςΨΏΓΔ…η±ΗΓΔ≤ΡΝœΦΑΤδΥϋΈοΤΖΘ§Έό¬έ «ΒΞΕά Ι”ΟΜΙ «ΉιΚœ Ι”ΟΘ§»γ–η“ΣΑϋά®»μΦΰΒ»ΘΜ÷Μ“Σ…η±ΗΤδ «’κΕ‘»ΥΧεΨΏ±Η“‘œ¬“Μ–©ΡΩΒΡΒΡ«ΑΧαœ¬:
’οΕœΓΔ‘ΛΖάΓΔΦύ≤βΓΔ÷ΈΝΤΜρΜΚΫβΦ≤≤Γ,
’οΕœΓΔΦύ≤βΓΔ÷ΈΝΤΓΔΜΚΫβΜρ≤Ι≥Ξ ή…ΥΜρ≤–Φ≤,
Βς≤ιΘ§ΗϋΜΜΜρ–όΗΡΫβΤ Μρ…ζάμΙΐ≥ΧΒΡ,
ή‘–ΩΊ÷Τ,
2:»γΚΈΕ®“ε≤ζΤΖΖ÷άύ
“ΫΝΤ…η±ΗΖ÷”–≤ΜΆ§ΒΡάύ±πΒ»ΦΕΗυΨί“ΫΝΤ…η±Η÷ΗΝν÷ΗΒΦ–‘ΈΡΦΰΘ®MEDDEV 2.4)≤Δ«“ΟΩΗω÷ΗΝν”–“ΜΗωΙφ‘ρΘ®RuleΘ©
Class I other “ΜάύΤδΥϊ
Class I sterile “ΜάύΟπΨζ
Class I measurement function “Μάύ≤βΝΩ
Class IIa 2aάύ
Class IIb 2bάύ
Class III and Class III with medicine »ΐάύΦΑ»ΐάύ¥χ“©Έο
3:»γΚΈΕ®“ε≤ζΤΖΖ÷άύ
Ήœ»≤ζΤΖΒΡΙφ‘ρ «»ΖΕ®ΒΡ
Τδ¥ΈΙφ‘ρœ‘ ΨΝΥ≤ζΤΖΒΡΒ»ΦΕΖ÷άύ
ΒΎ»ΐΖϊΚœ–‘ΤάΙάΒΡΡΘ Ϋ «±Μ―Γ‘ώΒΡ
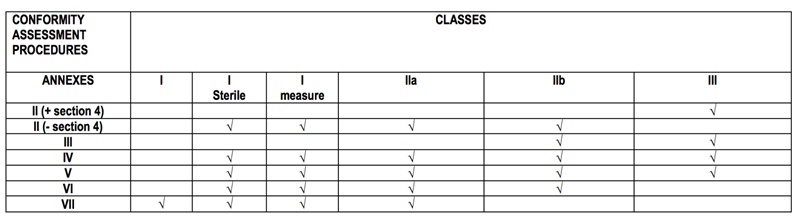
4:»œ÷ΛΝς≥Χ
Υυ”–ΒΡΖϊΚœ–‘ΤάΙάΕΦ Φ”Ύ“‘œ¬ΘΚ
1) ’ΒΫ…ξ«κ±μ
2) «© πΚœΆ§
3) ΦΤΜ°…σ≥ß
4) Β ©…σ≥ß
5) «©ΖΔ÷Λ ι / ÷Ί…σΚΥ /Β»¥ΐNC≤ΜΖϊœνΙΊœν
5:ΖϊΚœ–‘ΤάΙά≥Χ–ρ
Ζ÷άύ“άΨίMDD÷ΗΝνΗΫ¬ΦΨ≈
2. ―Γ‘ώ“ΜΗωΖϊΚœ–‘ΤάΙά≥Χ–ρ
3. ΙΪΗφΜζΙΙ÷¥––ΖϊΚœ–‘ΤάΙά
(Χα…ξ«κ, …σΚΥΦΦ θΈΡΦΰ, œ÷≥Γ…σΚΥ»»)
4. ΙΪΗφΜζΙΙ«©ΖΔ÷Λ ι (÷Λ ι”––ßΤΎ5Ρξ)
5. Ρξ…σ
(≥θ¥Έ…σΚΥ÷°ΚσΒΡ1Ρξ“ΜΕ®“ΣΫχ––Ρξ…σ.)
(ΙΪΗφΜζΙΙΟΩ»ΐΡξ“Μ¥ΈΆΜΜς…σΚΥΘ§ ±ΦδΩ…“‘ΗϋΕΧ.)
6:MDR…ξ«κ(’¬ΫΎ 3.1 ®C ΕΈ¬δ 1)
ΡβΕ®ΒΡ–¬Ζ®ΙφΚή¥σ≥ΧΕ»…œά©’ΙΚ≠Η«ΝΥ90/384/EEC÷ΗΝν“‘ΦΑ93/42/EEC÷ΗΝνΘΜάΐ»γ ΘΚΥϋΑϋά®ΝΥΥυ”–≥ΐΝΥΧεΆβ’οΕœ…η±ΗΒΡ“ΫΝΤΤς–ΒΓΘ
ΝμΆβΘ§≥ΐ¥Υ÷°ΆβΘ§ΖΕΈßΜΙά©’ΙΑϋά®ΝΥ“Μ–©œ÷”–÷ΗΝνAIMDD/MDDΟΜ”–ΑϋΚ§ΒΡ≤ζΤΖΓΘΜΙ”–“Μ–©≤ζΤΖ «‘Ύ“Μ–©≥…‘±Ιζ±Μ»œΈΣ «“ΫΝΤΤς–Β»¥≤Μ‘Ύ÷ΗΝνΖΕΈßΡΎΒΡΓΘ
7:MDR…ξ«κ
ά©’ΙΒΡΖΕΈß÷ς“ΣΑϋά®ΝΥ:
÷Τ‘λ≤…”ΟΖ«Ω…––ΒΡ»ΥΧεΉι÷·ΜρœΗΑϊΒΡ≤ζΤΖΘ§ΜρΤδ―ή…ζΈοΘ§“―Ψ≠ΖΔ…ζΝΥ Β÷ –‘ΒΡ≤ΌΉςΘ®άΐ»γΉΔ…δΤς‘ΛΉΑ”κ»ΥΧεΫΚ‘≠ΒΑΑΉΘ©Θ§≥ΐΖ«ΥϊΟ«±ΜΖ®ΙφΘ®ECΘ©1394 / 2007…œœ»ΫχΒΡ÷ΈΝΤ“©”Ο≤ζΤΖΥυΚ≠Η«ΓΘ
»ΥΧεΉι÷·ΚΆœΗΑϊΘ§Μρ≤ζΤΖά¥Ή‘»ΥΧεΉι÷·ΜρœΗΑϊΘ§Μυ±Ψ…œΟΜ”–≤ΌΉίΘ§ ή÷ΗΝν2004Θ·23Θ·EC≈ΖΟΥ“ιΜαΚΆ2004Ρξ3‘¬31»’÷ΤΕ®÷ ΝΩ±ξΉΦΚΆΑ≤»ΪΒΡΨη‘υΓΔ≤…ΙΚΓΔΦλ―ιΓΔΦ”ΙΛΓΔ±Θ¥φΈ·‘±ΜαΓΔ»ΥΧεΉι÷·ΚΆœΗΑϊ¥φ¥ΔΚΆΖ÷≤Φ≤Μ ήΗΟΖΫΑΗΙήΩΊ
Ρ≥–©÷≤»κ ΫΜρΤδΥϊ«÷»κ ΫΒΡ≤ζΤΖΟΜ”–“ΫΝΤΒΡΡΩΒΡ»¥άύΥΤ”Ύ“ΫΝΤ…η±ΗΒΡΧΊΒψΚΆΖγœ’Ή¥ΩωΘ®άΐ»γΖ«ΫΟ’ΐ–‘“ΰ–Έ―έΨΒΘ§ΟάΆΪΘ©
8:MDR Ε®“ε
“ΫΝΤ…η±ΗΓ· “βΈΕΉ≈»ΈΚΈ“«ΤςΓΔΤς–ΒΓΔ»μΦΰΓΔ÷≤»κΤΖΓΔ ‘ΦΝΓΔ≤ΡΝœΜρΤδΥϊΈοΦΰΘ§Έό¬έΒΞΕά Ι”ΟΜΙ «ΉιΚœ Ι”ΟΘΜ÷Τ‘λ Ι”ΟΡΩΒΡ «ΘΚΕ‘»ΥΧε≤ζ…ζ“Μ÷÷ΜρΕύ÷÷“ΫΝΤΡΩ±ξ÷ν»γΘΚ
®C’οΕœΓΔ‘ΛΖάΓΔΦύ≤βΓΔ÷ΈΝΤΜρΜΚΫβΦ≤≤Γ
®C’οΕœΓΔΦύ≤βΓΔ÷ΈΝΤΓΔΜΚΫβΜρ≤Ι≥Ξ ή…ΥΜρ≤–Φ≤
®CΒς≤ιΘ§ΗϋΜΜΜρ–όΗΡΒΡΫβΤ Μρ…ζάμΙΐ≥ΧΜρΉ¥Χ§
®C ή‘–÷ß≥÷Μρ’ΏΩΊ÷Τ
®C»ΈΚΈ…œ ω≤ζΤΖœϊΕΨΜρΟπΨζ
9:±δΗϋ ±Φδ
4.png
10:Ζ÷άύΙφ‘ρ
Ζ÷άύΙφ‘ρ
Rule 1~4 Ζ««÷»κ–‘…η±Η
Rule 5~8 «÷»κ–‘…η±Η
Rule 9~12 ”–‘¥…η±Η
Rule 12~18 ΧΊ βΙφ‘ρ
±δΗϋ
Rule 3 ®C ή‘–÷ß≥÷
Rule 6 ®C »Ξ≥ΐΩ…÷ΊΗ¥ Ι”Ο ÷ θΤς–Β
Rule 8 ®CAIMD‘ωΦ”“Μ–©ΑϋΚ§‘ΎAIMD÷ΗΝνœ¬≤ζΤΖ
Rule 9 ®C AIMD÷ΗΝνΡΎ»ίΒΡ‘ωΦ”
Rule 16 ‘ωΦ”¥ ΜψΓΑΚΥ¥≈Ι≤’ώΓΔ≥§…υ≤®Γ±
Rule 17 ®C »ΥΧε‘¥ΜρΕ·Έο‘¥…η±Η
Rule 19~21 ®C –¬Ιφ‘ρ
1: ≤Ο¥ «93/42/EEC“ΫΝΤΤς–Β÷ΗΝν?
÷ΗΝνΑϋά®ΝΥ“ΫΝΤ…η±Η“‘ΦΑΥϋΒΡ≈δΦΰ
ΗυΨί÷ΗΝν“ΫΝΤΤς–Β…η±Η“βΈΕΉ≈:
»ΈΚΈ“«ΤςΓΔΤςΨΏΓΔ…η±ΗΓΔ≤ΡΝœΦΑΤδΥϋΈοΤΖΘ§Έό¬έ «ΒΞΕά Ι”ΟΜΙ «ΉιΚœ Ι”ΟΘ§»γ–η“ΣΑϋά®»μΦΰΒ»ΘΜ÷Μ“Σ…η±ΗΤδ «’κΕ‘»ΥΧεΨΏ±Η“‘œ¬“Μ–©ΡΩΒΡΒΡ«ΑΧαœ¬:
’οΕœΓΔ‘ΛΖάΓΔΦύ≤βΓΔ÷ΈΝΤΜρΜΚΫβΦ≤≤Γ,
’οΕœΓΔΦύ≤βΓΔ÷ΈΝΤΓΔΜΚΫβΜρ≤Ι≥Ξ ή…ΥΜρ≤–Φ≤,
Βς≤ιΘ§ΗϋΜΜΜρ–όΗΡΫβΤ Μρ…ζάμΙΐ≥ΧΒΡ,
ή‘–ΩΊ÷Τ,
2:»γΚΈΕ®“ε≤ζΤΖΖ÷άύ
“ΫΝΤ…η±ΗΖ÷”–≤ΜΆ§ΒΡάύ±πΒ»ΦΕΗυΨί“ΫΝΤ…η±Η÷ΗΝν÷ΗΒΦ–‘ΈΡΦΰΘ®MEDDEV 2.4)≤Δ«“ΟΩΗω÷ΗΝν”–“ΜΗωΙφ‘ρΘ®RuleΘ©
Class I other “ΜάύΤδΥϊ
Class I sterile “ΜάύΟπΨζ
Class I measurement function “Μάύ≤βΝΩ
Class IIa 2aάύ
Class IIb 2bάύ
Class III and Class III with medicine »ΐάύΦΑ»ΐάύ¥χ“©Έο
3:»γΚΈΕ®“ε≤ζΤΖΖ÷άύ
Ήœ»≤ζΤΖΒΡΙφ‘ρ «»ΖΕ®ΒΡ
Τδ¥ΈΙφ‘ρœ‘ ΨΝΥ≤ζΤΖΒΡΒ»ΦΕΖ÷άύ
ΒΎ»ΐΖϊΚœ–‘ΤάΙάΒΡΡΘ Ϋ «±Μ―Γ‘ώΒΡ
4:»œ÷ΛΝς≥Χ
Υυ”–ΒΡΖϊΚœ–‘ΤάΙάΕΦ Φ”Ύ“‘œ¬ΘΚ
1) ’ΒΫ…ξ«κ±μ
2) «© πΚœΆ§
3) ΦΤΜ°…σ≥ß
4) Β ©…σ≥ß
5) «©ΖΔ÷Λ ι / ÷Ί…σΚΥ /Β»¥ΐNC≤ΜΖϊœνΙΊœν
5:ΖϊΚœ–‘ΤάΙά≥Χ–ρ
Ζ÷άύ“άΨίMDD÷ΗΝνΗΫ¬ΦΨ≈
2. ―Γ‘ώ“ΜΗωΖϊΚœ–‘ΤάΙά≥Χ–ρ
3. ΙΪΗφΜζΙΙ÷¥––ΖϊΚœ–‘ΤάΙά
(Χα…ξ«κ, …σΚΥΦΦ θΈΡΦΰ, œ÷≥Γ…σΚΥ»»)
4. ΙΪΗφΜζΙΙ«©ΖΔ÷Λ ι (÷Λ ι”––ßΤΎ5Ρξ)
5. Ρξ…σ
(≥θ¥Έ…σΚΥ÷°ΚσΒΡ1Ρξ“ΜΕ®“ΣΫχ––Ρξ…σ.)
(ΙΪΗφΜζΙΙΟΩ»ΐΡξ“Μ¥ΈΆΜΜς…σΚΥΘ§ ±ΦδΩ…“‘ΗϋΕΧ.)
6:MDR…ξ«κ(’¬ΫΎ 3.1 ®C ΕΈ¬δ 1)
ΡβΕ®ΒΡ–¬Ζ®ΙφΚή¥σ≥ΧΕ»…œά©’ΙΚ≠Η«ΝΥ90/384/EEC÷ΗΝν“‘ΦΑ93/42/EEC÷ΗΝνΘΜάΐ»γ ΘΚΥϋΑϋά®ΝΥΥυ”–≥ΐΝΥΧεΆβ’οΕœ…η±ΗΒΡ“ΫΝΤΤς–ΒΓΘ
ΝμΆβΘ§≥ΐ¥Υ÷°ΆβΘ§ΖΕΈßΜΙά©’ΙΑϋά®ΝΥ“Μ–©œ÷”–÷ΗΝνAIMDD/MDDΟΜ”–ΑϋΚ§ΒΡ≤ζΤΖΓΘΜΙ”–“Μ–©≤ζΤΖ «‘Ύ“Μ–©≥…‘±Ιζ±Μ»œΈΣ «“ΫΝΤΤς–Β»¥≤Μ‘Ύ÷ΗΝνΖΕΈßΡΎΒΡΓΘ
7:MDR…ξ«κ
ά©’ΙΒΡΖΕΈß÷ς“ΣΑϋά®ΝΥ:
÷Τ‘λ≤…”ΟΖ«Ω…––ΒΡ»ΥΧεΉι÷·ΜρœΗΑϊΒΡ≤ζΤΖΘ§ΜρΤδ―ή…ζΈοΘ§“―Ψ≠ΖΔ…ζΝΥ Β÷ –‘ΒΡ≤ΌΉςΘ®άΐ»γΉΔ…δΤς‘ΛΉΑ”κ»ΥΧεΫΚ‘≠ΒΑΑΉΘ©Θ§≥ΐΖ«ΥϊΟ«±ΜΖ®ΙφΘ®ECΘ©1394 / 2007…œœ»ΫχΒΡ÷ΈΝΤ“©”Ο≤ζΤΖΥυΚ≠Η«ΓΘ
»ΥΧεΉι÷·ΚΆœΗΑϊΘ§Μρ≤ζΤΖά¥Ή‘»ΥΧεΉι÷·ΜρœΗΑϊΘ§Μυ±Ψ…œΟΜ”–≤ΌΉίΘ§ ή÷ΗΝν2004Θ·23Θ·EC≈ΖΟΥ“ιΜαΚΆ2004Ρξ3‘¬31»’÷ΤΕ®÷ ΝΩ±ξΉΦΚΆΑ≤»ΪΒΡΨη‘υΓΔ≤…ΙΚΓΔΦλ―ιΓΔΦ”ΙΛΓΔ±Θ¥φΈ·‘±ΜαΓΔ»ΥΧεΉι÷·ΚΆœΗΑϊ¥φ¥ΔΚΆΖ÷≤Φ≤Μ ήΗΟΖΫΑΗΙήΩΊ
Ρ≥–©÷≤»κ ΫΜρΤδΥϊ«÷»κ ΫΒΡ≤ζΤΖΟΜ”–“ΫΝΤΒΡΡΩΒΡ»¥άύΥΤ”Ύ“ΫΝΤ…η±ΗΒΡΧΊΒψΚΆΖγœ’Ή¥ΩωΘ®άΐ»γΖ«ΫΟ’ΐ–‘“ΰ–Έ―έΨΒΘ§ΟάΆΪΘ©
8:MDR Ε®“ε
“ΫΝΤ…η±ΗΓ· “βΈΕΉ≈»ΈΚΈ“«ΤςΓΔΤς–ΒΓΔ»μΦΰΓΔ÷≤»κΤΖΓΔ ‘ΦΝΓΔ≤ΡΝœΜρΤδΥϊΈοΦΰΘ§Έό¬έΒΞΕά Ι”ΟΜΙ «ΉιΚœ Ι”ΟΘΜ÷Τ‘λ Ι”ΟΡΩΒΡ «ΘΚΕ‘»ΥΧε≤ζ…ζ“Μ÷÷ΜρΕύ÷÷“ΫΝΤΡΩ±ξ÷ν»γΘΚ
®C’οΕœΓΔ‘ΛΖάΓΔΦύ≤βΓΔ÷ΈΝΤΜρΜΚΫβΦ≤≤Γ
®C’οΕœΓΔΦύ≤βΓΔ÷ΈΝΤΓΔΜΚΫβΜρ≤Ι≥Ξ ή…ΥΜρ≤–Φ≤
®CΒς≤ιΘ§ΗϋΜΜΜρ–όΗΡΒΡΫβΤ Μρ…ζάμΙΐ≥ΧΜρΉ¥Χ§
®C ή‘–÷ß≥÷Μρ’ΏΩΊ÷Τ
®C»ΈΚΈ…œ ω≤ζΤΖœϊΕΨΜρΟπΨζ
9:±δΗϋ ±Φδ
4.png
10:Ζ÷άύΙφ‘ρ
Ζ÷άύΙφ‘ρ
Rule 1~4 Ζ««÷»κ–‘…η±Η
Rule 5~8 «÷»κ–‘…η±Η
Rule 9~12 ”–‘¥…η±Η
Rule 12~18 ΧΊ βΙφ‘ρ
±δΗϋ
Rule 3 ®C ή‘–÷ß≥÷
Rule 6 ®C »Ξ≥ΐΩ…÷ΊΗ¥ Ι”Ο ÷ θΤς–Β
Rule 8 ®CAIMD‘ωΦ”“Μ–©ΑϋΚ§‘ΎAIMD÷ΗΝνœ¬≤ζΤΖ
Rule 9 ®C AIMD÷ΗΝνΡΎ»ίΒΡ‘ωΦ”
Rule 16 ‘ωΦ”¥ ΜψΓΑΚΥ¥≈Ι≤’ώΓΔ≥§…υ≤®Γ±
Rule 17 ®C »ΥΧε‘¥ΜρΕ·Έο‘¥…η±Η
Rule 19~21 ®C –¬Ιφ‘ρ